# Install BiocParallel if needed
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("BiocParallel")26 Parallelizing Workflows
26.1 What is parallelization?
Parallelization refers to the process of dividing a computational task into smaller sub-tasks that can be executed simultaneously (in parallel) across multiple processing units, such as CPU cores, GPUs, or even distributed systems. The goal is to speed up computations by taking advantage of modern hardware’s ability.
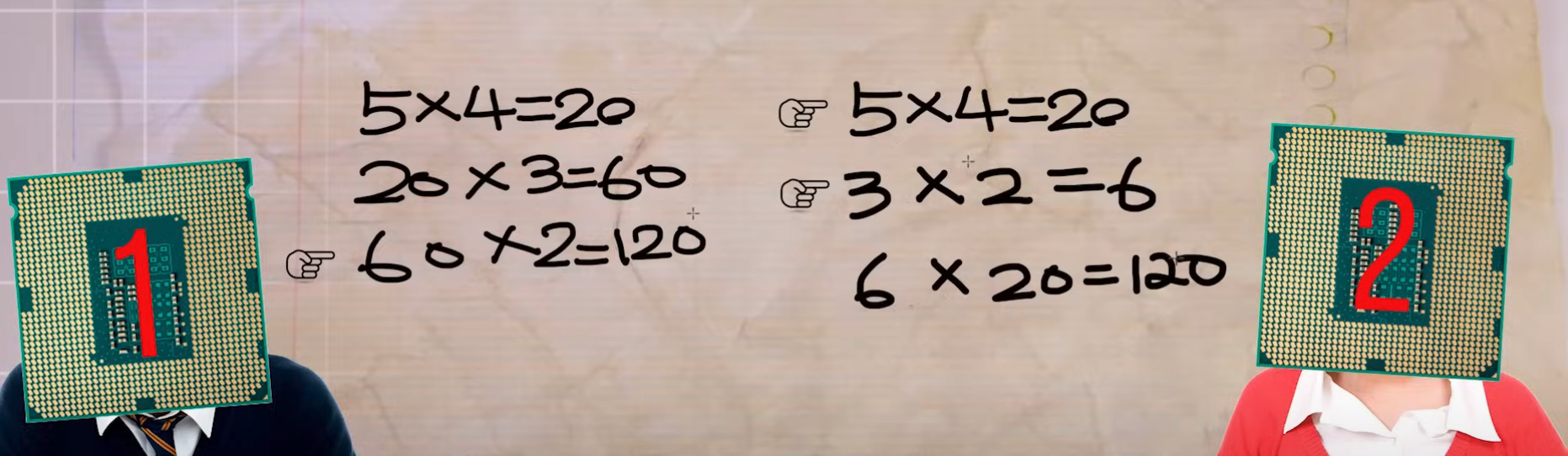
26.2 Examples of parallelization
- Data Parallelism: Dividing large datasets into smaller chunks and processing them simultaneously using the same operation.
# Example
Aligning sequencing reads to a reference genome using tools like BWA or Bowtie,
can be parallelized by splitting the input FASTQ file into smaller chunks and
processing them on different CPU cores.- Task Parallelism: Different tasks are run in parallel, where each task performs a different operation. Jobs are independent. Alternatively the same task is run on different samples within your dataset.
# Example 1
Task 1: Run FastQC for quality control
Task 2: Align reads to the genome using STAR# Example 2
Run FastQC on each sample in parallel.- Pipeline Parallelism:: Breaking a process into stages, with each stage operating in parallel. Tasks are dependent.
# Example: RNA-seq pipeline
Stage 1: | Trim Batch A | Trim Batch B | Trim Batch C |
Stage 2: | Align Batch A | Align Batch B |
Stage 3: | Count Batch A |To learn more about parallelizing across samples, see Chapter 14.
26.3 Multi-core vs -process vs -thread?
26.3.0.1 What is CPU, core and thread?
- CPU: A kitchen.
- Core: A chef in the kitchen (physical worker).
- Thread: A recipe that the chef is following (task instructions).
26.3.0.2 When to use what strategy?
Multicore: Use for tools like BWA, STAR, or SAMtools, which explicitly support threading for intensive computations.
Multiprocess: Use when tasks are independent and require fault isolation or need to scale across machines. e.g. multiple windows/operations
Multithread: Use for I/O-heavy tasks like reading FASTQ files, downloading data, or parsing JSON. e.g. memory leak can be effected.
26.4 Tools for Parallelization in R or Python:
- R:
BiocParallel, future, future.batchtools - Python: multiprocessing, Dask, Ray
26.4.1 BiocParallel
BiocParallel aims to provide a unified interface to existing parallel infrastructure where code can be easily executed in different environments.
- STEP 1. Set up the Params
- STEP 2. Build a Function allowing Parallelization
- STEP 3. Check the state of the parallel evaluation env
- STEP 4. Error handling and logging
26.5 BiocParallel strategies
support different Parallelization strategies (e.g. multicore, snowparam ,,,) with a unified interface:
- SerialParam()
- SnowParam()
- MulticoreParam()
# load library
library("BiocParallel")
# Params
registered()
bpparam() # current param26.6 BiocParallel - bplapply(), bpvec() and biterate()
Provide parallel list iteration bplapply, vectorized operations bpvec and file iteration bpiterate. Parallelization introduces overhead that can make simple tasks slower than their sequential counterparts, especially when the task is very lightweight
# bplapply
start.time <- Sys.time()
numbers <- list(1:10000000)
square_function <- function(x) x^2
# square_function <- function(x) sum(sqrt(x^2))
result <- bplapply(numbers, square_function) # Q why it takes longer when i use bplapply?
end.time <- Sys.time()
time.taken <- end.time - start.time
time.taken# bpvec
numbers <- 1:10
result <- bpvec(numbers, FUN = square_function)
print(result)26.7 BiocParallel - foreach()
support of foreach: looping constructs that supports parallel execution
library(foreach)
param <- MulticoreParam(workers = 4) # Use 4 workers (cores)
register(param)
numbers <- 1:10
# Use foreach to compute squares in parallel with BiocParallel
result <- foreach(x = numbers, .combine = c, .options.BP = list(BPPARAM = param)) %do%
bplapply(list(x), function(x) x^2)
result26.8 BiocParallel - Others
BiocParallel works seamlessly with many of the bioconductor packages (including DESeq2). But BiocParallel also provides a whole paradigm for parallelizing workflows in R that attempts to be “portable”. The big idea is that your work can be run on other platforms (which may have varying levels of parallel capability), and that if you used this to implement your work, then it should still be able to run after end users did some minor configuration. So in that sense, BiocParallel is really built for scientific programmers.
26.8.0.1 REFERENCE
26.9 future package
The future package provides a way to create ‘futures’, which helps with parallelism since those things can be evaluated asynchronously. Many other packages in the R world will use the futures package to implement parallelism, and so often you will toggle on a parallel way of working by just knowing about this. The seurat package provides a common example.
# single cell: Seurat
library(future)
plan("multisession", workers = 1)
plan()26.10 future.batchtools
What if you want to use the WHOLE cluster to do a really big number crunch, but you don’t really want to leave your R session?
Well in that case, you could make use of the future.batchtools package. This package combines the future package with the batchtools package to enable asynchronous parallelization on an HPC cluster. It’s compatible with slurm and (somewhat to my amazement) it works on our current cluster even with the use of containerized R sessions. Remarkable!
Using future.batchtools, you can write R code using standard looking methods (apply-like functions) and have the results quietly submitted to the cluster on your behalf while it is processing your code…
There is a small amount of set up in order to make this work though. Specifically, you need to provide the information needed by the scheduler in the form of a list that is passed to the resources argument to the plan() function as illustrated below:
library(future.apply)
library(future.batchtools)
## the call to plan() sets up the parameters for talking to the cluster (for each job)
plan(batchtools_slurm,
workers = 20,
resources = list(nodes = 1,
cpus_per_task = 1,
walltime = 180,
ntasks=1,
ncpus=1,
memory=1024,
account="cpu-test-sponsored",
partition="cpu-test-sponsored"),
template = "/data/hps/assoc/public/bioinformatics/templates/batchtools.slurm.tmpl")
## Then you actually give the command to tell the cluster to do work using an apply-like construct:
output <- future_sapply(1:100, function(i) mean(rnorm(1e7)), future.seed = 1)
## Finally, we can look at the output (after the job finishes)
outputNotice that one of the arguments provided is also a template. That template is the same for everyone on the system and it just helps the future.batchtools package to format things into slurm for you. To make this simple for everyone, we have just provided a working copy in the example above.
For more details on how to use the future.batchtools package, you might find this (https://computing.stat.berkeley.edu/tutorial-dask-future/R-future.html#1-overview-futures-and-the-r-future-package)[link] to be helpful.
26.11 future.batchtools and BiocParallel
future.batchtools is also supported as part of BiocParallel, so you will also see it referred to in the documentation there. Here is an example of how that might look:
## define work to be done
FUN <- function(i) system("hostname", intern=TRUE)
## load BiocParallel
library(BiocParallel)
## register SLURM cluster details and the template file
param <- BatchtoolsParam(workers=100,
cluster="slurm",
resources = list(nodes = 4,
cpus_per_task = 1,
walltime = 180,
ntasks=1,
ncpus=1,
memory=1024,
account="cpu-test-sponsored",
partition="cpu-test-sponsored"),
template="/data/hps/assoc/public/bioinformatics/templates/batchtools.slurm.tmpl")
register(param)
## do work
xx <- bplapply(1:100, FUN)
## look at the data we produced
table(unlist(xx))